Galaxy services
- Category: Web-based service for bioinfomatics analysis
- Coordination: support.sigenae@inrae.fr
- Service: since 2012
- Service URL: https://sigenae-workbench.toulouse.inrae.fr
Sigenae's Galaxy instance: from system administration to software engineering
Galaxy (https://galaxyproject.org) is an analysis
web interface whose development began in 2006-2007 with a first version distributed in mid 2010.
It is actively maintained by the "Galaxy project" team at Penn State, Johns Hopkins University,
Oregon Health & Science University for people who want to process (bio)computer data without specific computer skills.
The four main objectives of galaxy are: accessible, reproductible, transparent genomic science, and community centered.
An active community around Galaxy develop tools and share within the Galaxy ToolShed.
Galaxy can be used by researchers in different ways : on their personnal computer, or via remote web servers.
A free public web server Galaxy is provided by the Galaxy Project, but for production environment with huge amount of data and heavy computations,
the best is to setup a local instance of this web service, closer to our local data storage et HPC cluster.
Sigenae team does not have its own IT infrastructure, but relies on that of Genotoul BioInfo and is involved in its system administration. Access to our local instance of Galaxy is given to any user of Genotoul BioInfo plateform having a valid LDAP account. Dedicated training Galaxy intances with local accounts can also be provided on demand.
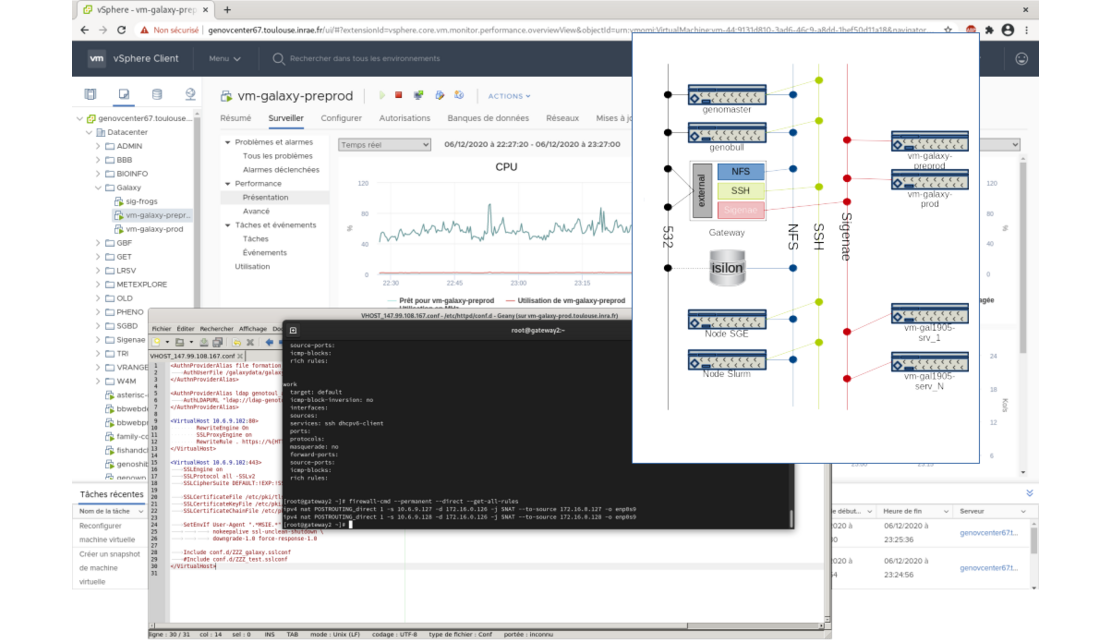
All these virtual machines are deployed on Genotoul BioInfo VMware virtualisation infrastructure.
This implies fine tuning skills in network administration, firewall rules definition, Web proxying, and VMware management of course.
These virtual machines only handle the web part of Galaxy, all the data processing is dispatched on Genotoul BioInfo HPC cluster using Slurm DRMAA.
Input and output data are located on Genotoul BioInfo storage facilities, and mounted on both cluster nodes and Galaxy virtual machines.
Genotoul BioInfo virtualisation infrastructure consists of 3 servers Dell PowerEdge R740 with 64 cores and 766,5GB each, connected to a 40 TB Dell Storage SCv3020.
Our "standard" Galaxy virtual machine is a 4 cores/16GB RAM one, with a 50GB local storage mainly used for PostgreSQL embedded Galaxy database which keep tracks of all analysis made by researchers and acts as a LIMS.
Several machines of this kind are deployed for production purpose and dedicated to some specific domains of bioinformatics analysis. Galaxy users' data is stored in a 40TB dedicated volume on the Genotoul BioInfo Isilon NFS storage clusters (1.6PB global capacity) with snapshots and replication facilities for data protection.
Boinformatics analysis are performed on 3 dedicated nodes Dell PowerEdge C6220 (40 cores with Hyper Threading/256 GB) of the Genotoul BioInfo Slurm HPC cluster composed of 68 PowerEdge C6220, 48 Bullx R424-E4 (64 cores HT/256 or 512GB) and two huge memory machines (1.5TB and 3TB). To speed up data access, cluster nodes are connected through EDR InfiniBand network to a 800TB Spectrum Scale storage cluster. Our Galaxy instance use this fast storage for job working directories.
Smaller "disposable" machines for training purpose are available with local accounts, and can be reset between training sessions to offer a new clean LIMS data environment.
All theses machines are on a private network and accessed through a secure web proxy.
System administration services outside Galaxy.
Many of the projects carried out by Sigenae have their deliverables presented via a website,
hosted on virtual machines managed by Sigenae.
Sigenae can also advise users on the IT infrastructure to be put in place to meet their needs,
or even set it up in collaboration with Genotoul BioInfo.
Started in 2012, Sigenae's Galaxy instance evolves with Genotoul BioInfo infrastructure for the hardware part. And try to follow Galaxy versions that provide major new functionalities.
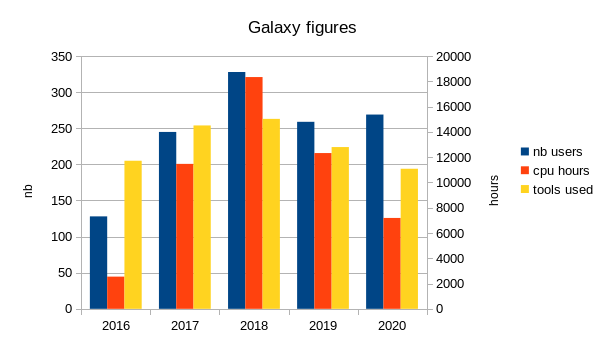
During last years, the number of users varies from year to year, but stabilizes around 250 if training is included, and around 160 if only those with more than one hour of true computing time are included.
The number of computing hours per year done via our Galaxy instance is on average 10000 hours.
Each year, arround 200 different tools are used to process data.
Software development often responds to specific needs encountered during analysis projects among our users.
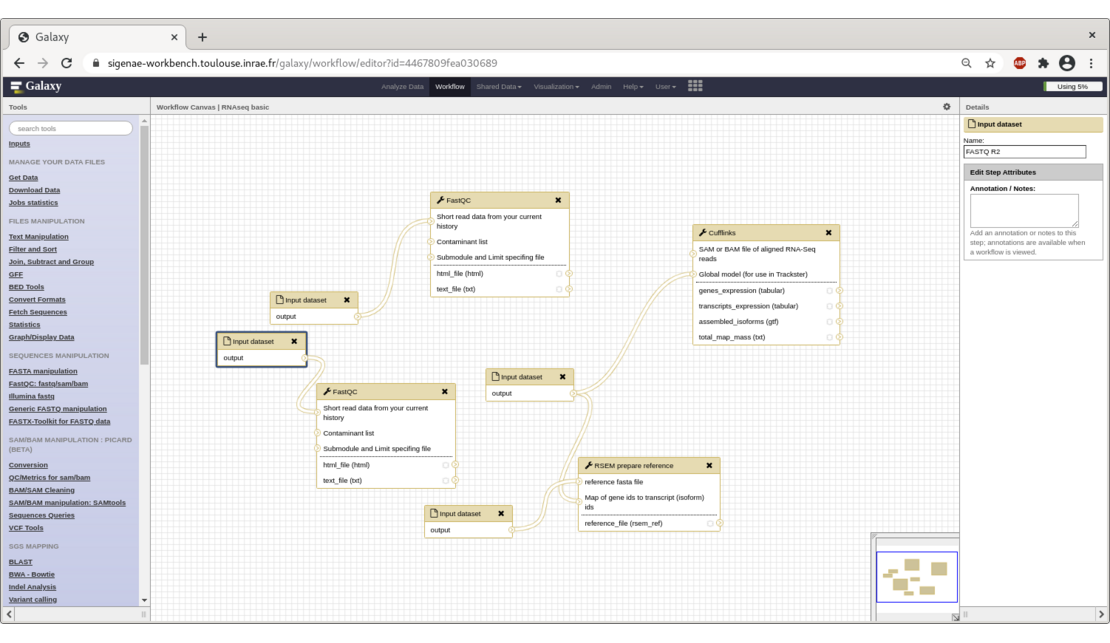
Doing bioinformatics analysis in Galaxy requires tools, sort of script written in XML/Cheetah that defines inputs and outputs, the chaining of command lines that will execute software, but also the web form design used for parameters specification. A software can't be used directly inside Galaxy without such a script called its wrapper tool.
Although many tools are available in the Galaxy toolshed (public tools repository), we wrote wrapper for "too" new software, or even developed some to meet the specific needs of our users :
-
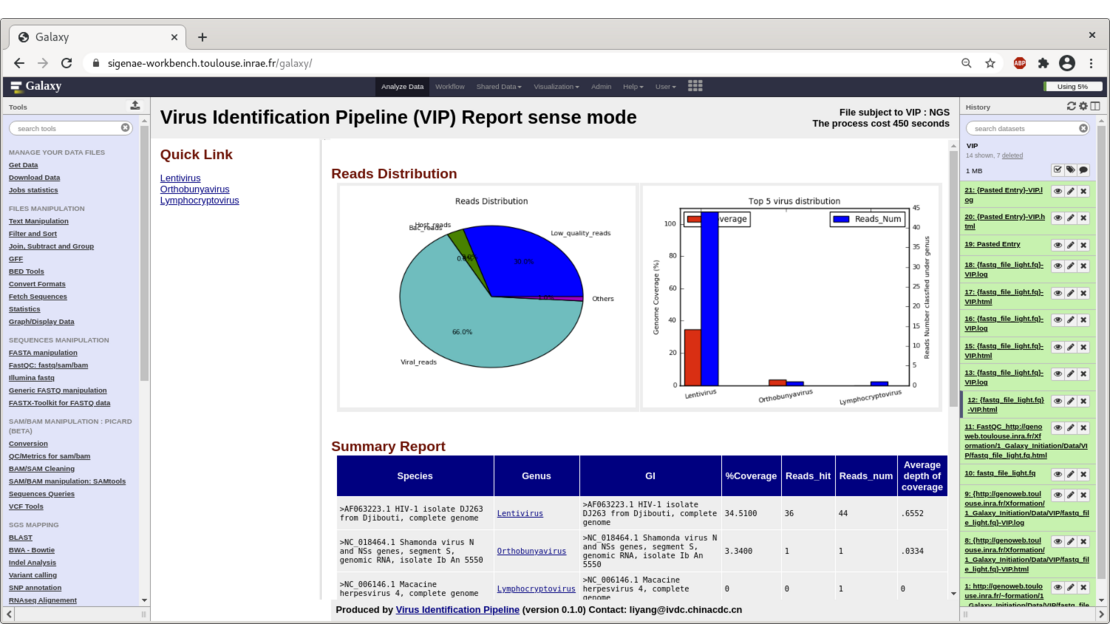
VIP detects viral sequences for the treatment of NGS data from viral metagenomics,
Tax4Fun predicts the functional or metabolic capabilities of microbial communities based on 16S data samples,
MirDeep2 mapper, prediction and quantification tools have been wrapped in Galaxy for sRNAseq analysis,
Some SNP tools (Variant effect predictor, Protseq, Netphos, Mitoprot, gpi, netCglyc, netNglyc, netOglyc)
have been wrapped to analyse variants.
Some RNAseq tools have also been wrapped : htseq count, FeatreCounts, RSEM.
And other specifics tools to upload and download data to optimize Galaxy work space by creating symlinks.
To help our users explore their data, we provide them several ready-to-use workflows for SNPs, viral (VIP) and metagenomic analysis.
Workflows are not the exclusive domain of Galaxy, other wokflows managers exist such as Snakemake or Nextflow and are used within Sigenae, either as simple users or as developers. The latter are usually command line oriented, which can put off some users. The web interface aspect of Galaxy is a real plus, but the main contribution of Galaxy remains its LIMS side.
Sigenae software engineering: a question of granularity.
Software development at Sigenae is goal-driven. If software building blocks already exist to conduct bioinformatics analysis,
a workflow type development will be favored. If there are missing software building blocks,
they will be developed in the language best adapted to the type of processing.
In some cases, we rewrite a software solution for performance reasons. Optimization can simply consist of a change of programming language,
better input/output management, use of databases, parallelization by splitting the input data files into sub batches if possible,
or by fine parallelization (multi processing, multi threading).
Sigenae's role is therefore not limited to bioinformatics analysis and presentation of results, the team is able to develop and implement complete IT solutions to meet the needs of the researcher.